Week 05 Lecture: Protein Structure Prediction in the Age of Artificial Intelligence
Contents
Week 05 Lecture: Protein Structure Prediction in the Age of Artificial Intelligence¶
High-accuracy structural biology at the intersection of machine learning and physical modeling.
Protein sequence - structure - function paradigm¶

Proteins with similar amino acid sequence have similar structure and similar function.
If we know the sequence, can we predict the structure?
If we know the structure, can we understand function?
Levels of protein structure¶

Secondary structure prediction¶
Neural networks were used early on for secondary structure prediction.

Neural networks were used early on for secondary structure prediction.

Weight matrices in early networks were simple.

Improved networks learn from sequence alignments.

Improved networks learn from sequence alignments (encoded with more sensitive position-specific iterated alignment matrices).

Secondary structure prediction accuracies routinely reached 80% by 2002 (CASP5).
PSIPRED server gives reliable answers in a few minutes.
http://bioinf.cs.ucl.ac.uk/psipred

Simulations can predict secondary structures as well, but with much more computational effort and results are very sensitive to the choice of force field.

Tertiary structure prediction¶
Tertiary protein structure prediction accuracy has improved dramatically in recent years.

Homology modeling¶
Structures are predicted based on sequence-structure similarity.
If we know structure for one protein, we can generate a good model for another protein with a similar sequence (e.g. Human thioredoxin based on E. coli). This requires good sequence alignment and model building tools to mutate side chains and add missing parts.
Improved protocols assemble structures from homologous and de novo fragments.
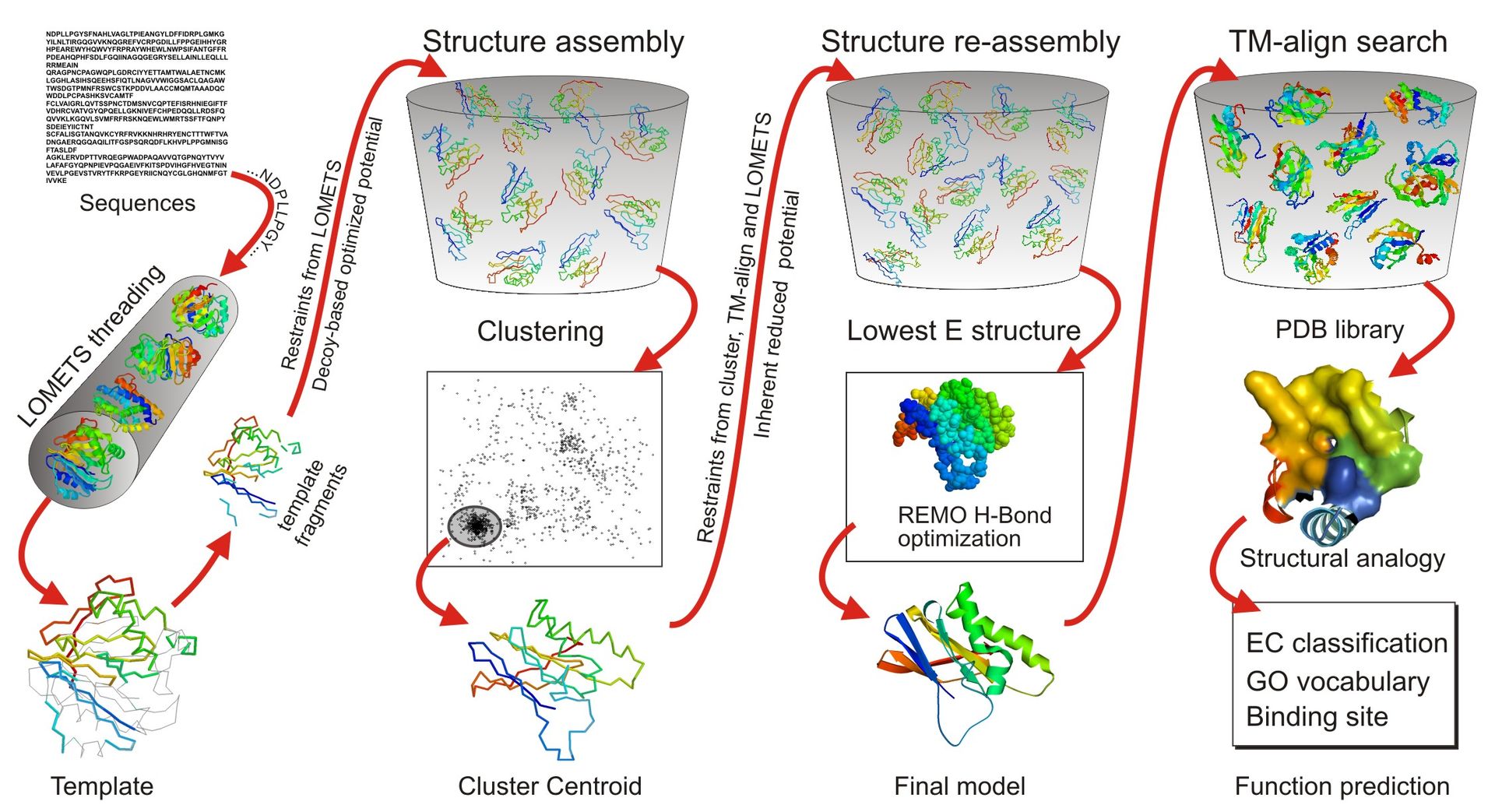
Physics-based protein folding¶
Molecular dynamics simulations can fold proteins, but with great computational effort.

Molecular dynamics simulations can also refine predictions.

Homology modeling without contact restraints¶
Modeling with contact restraints¶
Contact predictions via machine learning¶
Initial methods were based on statistical inference, but ML methods perform better.
AlphaFold 1¶
Combined distograms (and backbone torsion angles) predicted from coevolutionary couplings with generative models trained to maximize GDT similarity scores.

AlphaFold 1 generated actual structures either via a complex simulated annealing protocol or via gradient descent using potentials augmented by other potentials (Rosetta etc.)

trRosetta improves over AlphaFold1 by predicting residue distances and relative orientations

AlphaFold2¶
Introduction of attention-based transformer architecture with iterative refinement and directly generates structures (end-to-end predictions).

AlphaFold2’s ‘evoformer’ convolutes co-evolutionary information from the multiple sequence alignment with structural information (initially from templates, then iteratively from models that are generated). The architecture uses elements of graph neural networks to maintain equivariance.

AlphaFold2’s structure module directly generates structures via an attention-based module trained on actual structures but also augmented with physical constraints. Final models are generated after minimization with Amber (not shown).

AlphaFold2 models are remarkably accurate at the atomistic detail, including for side chains.

Errors are generally around 1Å RMSD but may be larger in loop regions or parts of a structure subject to contacts with other units, for example due to crystal packing.
Errors are generally around 1Å RMSD but may be larger in loop regions or parts of a structure subject to contacts with other units, for example due to crystal packing.

What happens when AlphaFold2 structures are used in simulations?

AlphaFold2 is very good at generating one structure, but what if there are multiple functional states?

RoseTTAfold is an alternative but conceptually similar approach to provide structures at accuracy approaching AlphaFold2.

Quaternary structure prediction¶
Predictions of complexes have remained a challenge, but much progress is being made:

Are we done?¶
“At present, for the best cases, the C-alpha coordinate RMSD accuracy of AlphaFold-predicted structures roughly corresponds to the accuracy expected for structures determined at resolutions no better than ∼4 Å. Thus, although structural predictions by AlphaFold and RoseTTAfold may be accurate enough to assist with experimental structure determination, they alone cannot provide the kind of detailed understanding of molecular and chemical interactions that is required for studies of molecular mechanisms and for structure-based drug design.”
and
“… solving the protein-folding problem means making accurate predictions of structures from amino acid sequences starting from first principles based on the underlying physics and chemistry.”